
Have a BLAST With Sequence Alignment - PArt 1

In biology, finding similarities enhances our understanding of the structures, organisms, or societies compared, allowing us to hypothesize function, decipher relationships, and construct ideas based on this information. Though sequence alignment and BLAST go deep into bioinformatics and computer work, they have a variety of uses all related to the goal of finding homologies in sequences (not to mention the fun and straightforward acronym, a rare phenomenon for most bioinformatics technologies)! In this article, we discuss what sequence alignment is, how it is used, and the different types.
In essence, sequence alignment aims to identify how much and in what ways two sequences are alike, positioning the sequences so that they obtain the highest number of similarities. These similarities are identified by a computer program and then separated by “gaps” to help align the matching nucleotides. However, when dealing with two vastly different species with a high frequency of mutations—deletions, insertions, or substitutions—in a sense, sequence alignment aims to minimize the differences.
There are three primary utilities of sequence alignment: functional, evolutionary, and structural similarities. Identifying similarities in function can lead to the discovery of putative functions and early-stage characteristics of certain genes. For example, finding a homology between mouse and human genes can allow researchers to identify what the novel gene may most closely act like steering researchers the right way when conducting further studies. From the perspective of evolution, sequence alignment can help to determine the relationships between species, allowing researchers to determine what genes have diverged over time and what mutations have accumulated. Finally, sequence alignment can be used to detect conserved structural motifs, such as a region, section, or domain, in different proteins or nucleic acids. Some examples include the zinc-binding domain, a regulator of protein function, the TATA box, a promoter sequence found in eukaryotes that indicate the start of transcription for a gene, or even used to identify a family of proteins, such as DNA binding proteins. However, it is important to note that identifying such similarities between sequences is not conclusive in the absence of experimental validation.
Though comparing two sequences with 10 base pairs each may seem somewhat straightforward when dealing with millions upon millions of base pairs, it may be more difficult or even not possible to find perfect alignments; the best match between sequences is not clear and is obscure. Consequently, sequence alignment takes two different approaches: local and global alignment. Local alignment identifies regions of similarities between sequences that are dissimilar overall. This is particularly useful for more complicated sequences or when trying to determine evolutionary relationships between different species. A global approach, on the other hand, aims to align the entire sequence, making it more useful for similar sequences of generally similar lengths.
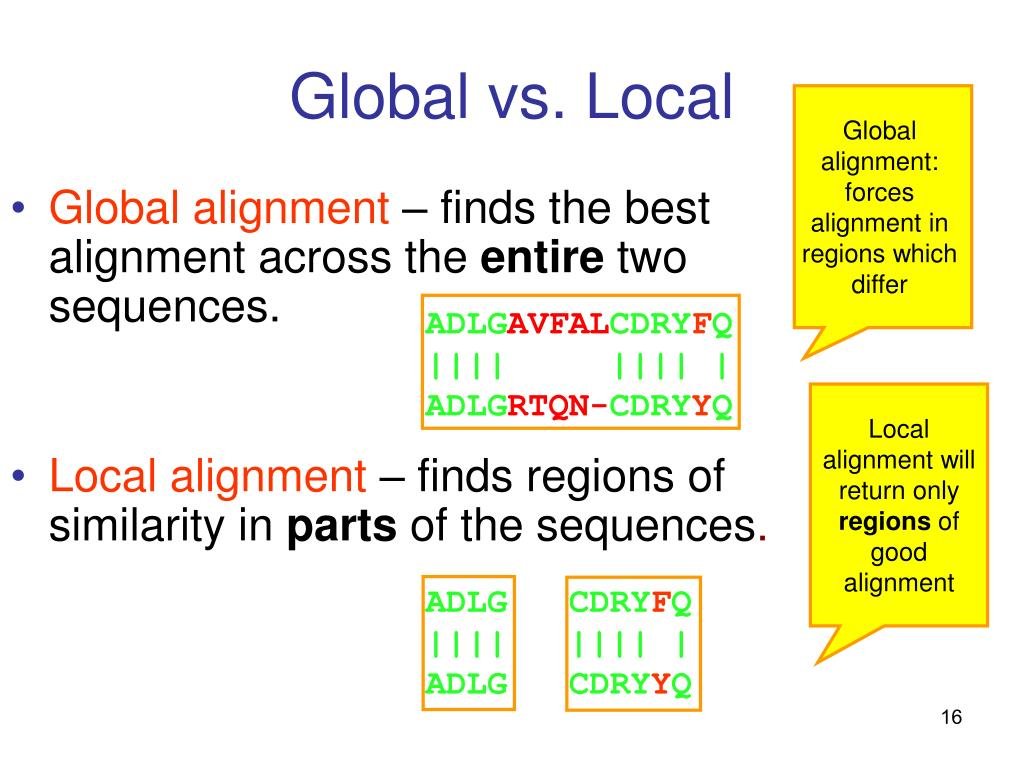
“Lesson 2.” SlideServe, Nov. 2014, www.slideserve.com/allistair-haley/lesson-2. Accessed 7 Aug. 2023.
There are three main methods or visualizations of the pairwise alignment data. The first is the dot-matrix method is a graphical alignment method that compares two sequences by placing dots on matching residues that are also used for aligned sequences, allowing us to see repetitive similar sequences. These dots indicate identical residues where the sequence is compared to itself, often forming a diagonal region. Other diagonals indicate similar or repetitive sequences.
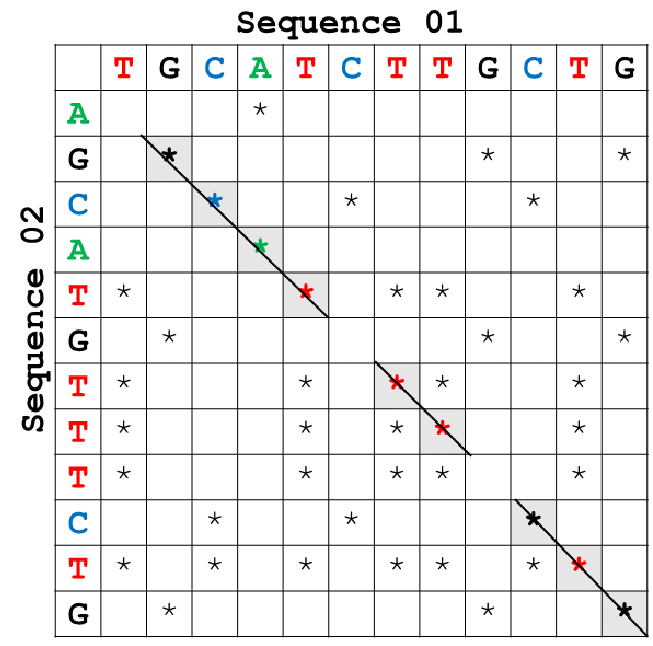
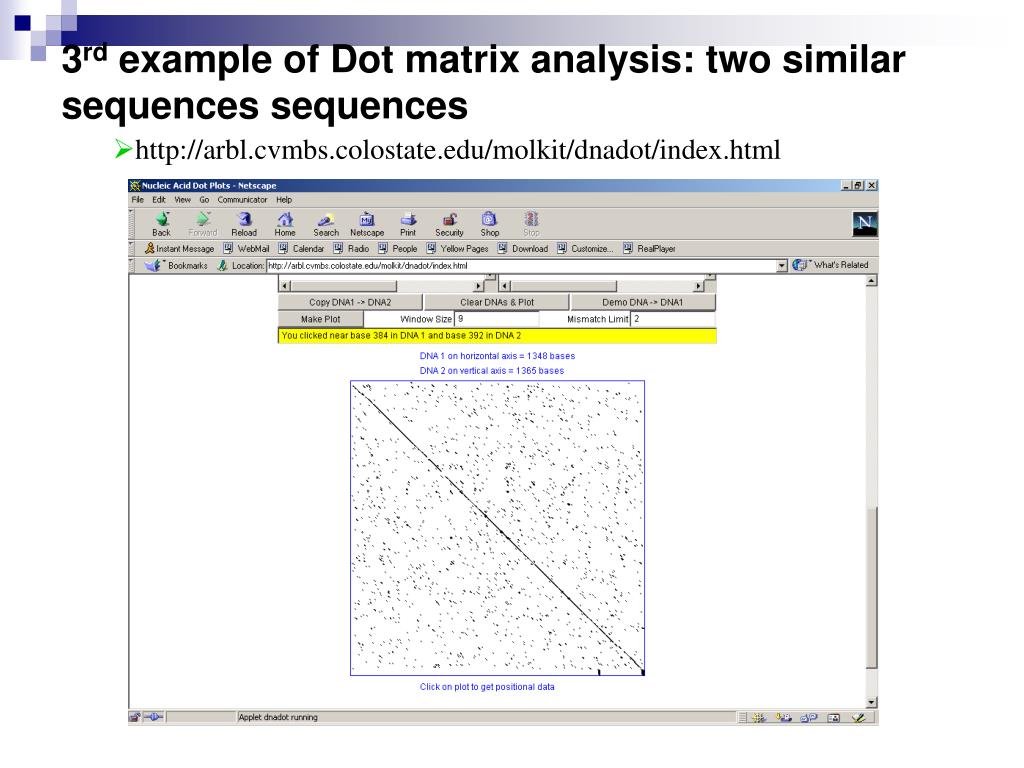

The next method, known as a dynamic sequence, compares all residue pairs using them to construct an optimal alignment. To identify the top alignment, scoring metrics such as the Smith-Waterman algorithm for local alignment or the Needleman-Wunsh algorithm for global alignment are employed to generate a score for similar residues and then use these scores to match the most similar sections of the sequences. The algorithm rewards itself for finding matches in similar residues and penalizing mismatches, depicted by the positive and negative numbers in the figures below. After running and trying to maximize its similarity score, it should reveal the most optimal path (indicated by arrows). The gaps and mismatches in the path may correspond to mutations.


Finally, the K-tuple method (also known as the Basic Local Alignment Search Tool method) uses short identical sequences in addition to alignment by dynamic programming. It starts by identifying similarities between just a few base pairs then builds the sequence from there, identifying more and more similarities. The algorithm enables the comparison of target sequences, nucleotides or proteins, with another sequence or database (such as the NCBI with millions of entries). In short, after receiving numerous amounts of information, BLAST identifies similarities between short stretches of nucleotides (as if it is comparing “words”) and then performs alignment. If a sequence is too long (perhaps over a couple of thousand base pairs), the efficacy of BLAST decreases. Still, despite this caveat, out of all the techniques discussed, BLAST is the method that is ubiquitously used in biology.

Some other special scenarios for alignment exist as well. With multiple sequences, the pairwise alignment methods discussed above can be used to compare them; however, multiple alignment methods must be used. The algorithms will often implement a hierarchical method to align the sequences, match the most similar pairs, then iterate through the less similar sequences until the full alignment is generated.
Another method is structural alignment. For proteins larger than RNAs, structural information can also be used to align a sequence, which is often more reliable since structure tends to be more conserved. The two methods of carrying out structural alignment include the Sequential Structural Alignment Program (SSAO) which compares atom to atom vectors (bonds between atoms) and distance matrix alignment (DALI) which breaks the protein into 6-peptide fragments and generates a distance matrix (similar to the word-based approach used in BLAST).
After understanding the mechanisms behind the BLAST and sequence alignment, now you have the chance to perform it yourself through this website: https://blast.ncbi.nlm.nih.gov/Blast.cgi. Visit the next blog to get a step-by-step run-through of this website, harnessing the full power to BLAST off into the world of bioinformatics!